





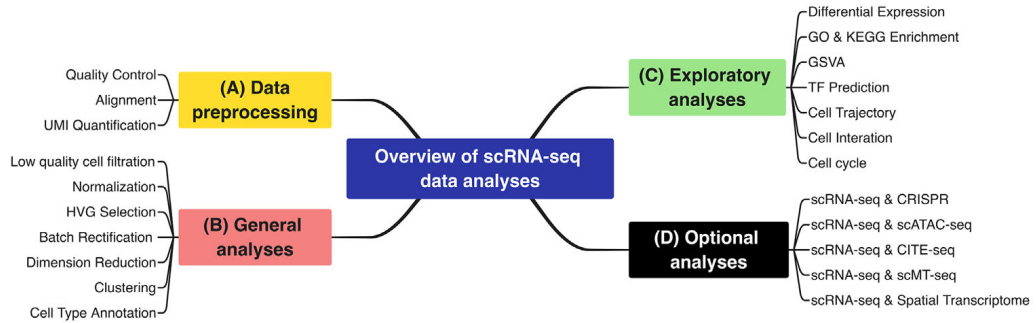
充分利用scRNA-seq数据
上两节我们介绍了用cellranger分析10x genomics单细胞转录组数据以及Smart-seq2测序数据的前期分析,获得了单细胞基因表达矩阵,那么接下来可以分析哪些内容呢?
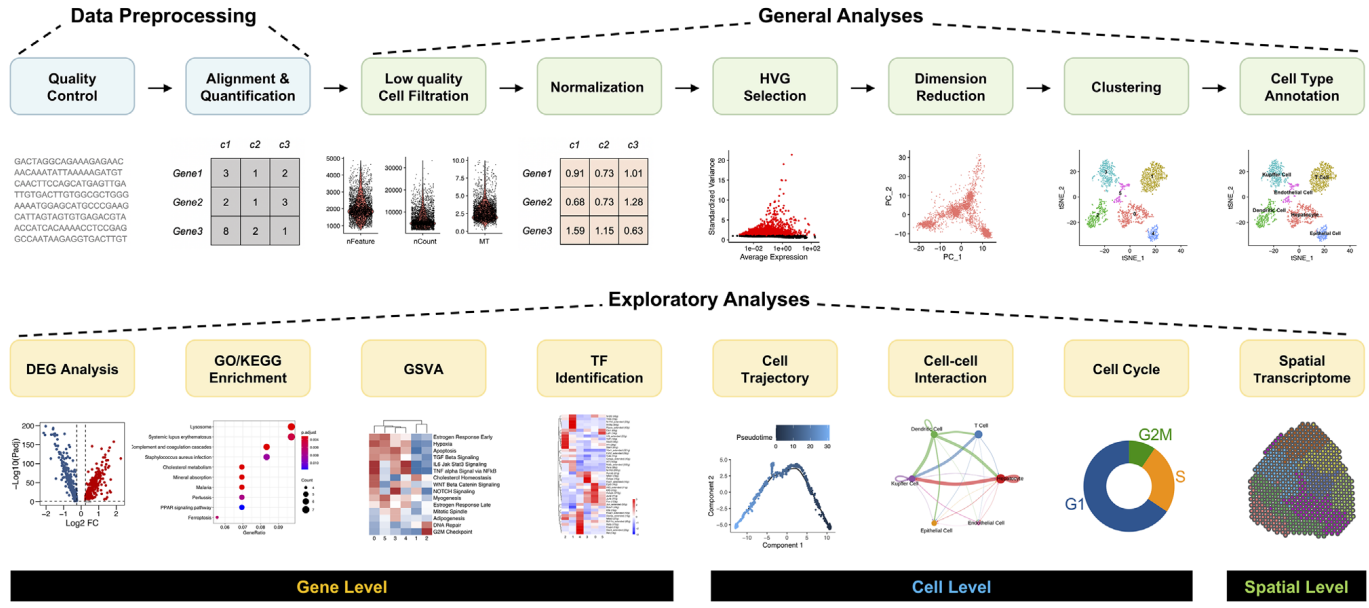
数据质控
在前期数据分析的时候,我们已经进行过一部分质控,但是那些质控主要是针对建库质量,如平均reads数、基因数、UMI数等以及测序质量,如低质量reads,接头序列等。但是在建库难免会引入死细胞、破裂细胞、或者发生细胞粘附导致的双细胞等现象,这就需要使用第三方软件如Seurat、Scanpy、Scran等进行分析。其中Seurat是最受欢迎的软件之一,可以根据UMI、基因数以及表达的细胞比例过滤不合格细胞或者基因等。
聚类注释
和bulk RNA-seq类似,在进行单细胞转录组分析时把每个细胞作为单独的个体。为了不同细胞之间可比,需要进行归一化处理,常见的归一化算法包括BASiCS, GRM, Linnorm, SAMstrt, SCnorm, scran和Simple Norm等。scRNA-seq数据是高维的,不同细胞中存在一群表达水平基本不变的基因(housekeeping gene),这类基因会掩盖真实的生物信息。所以需要鉴定高可变基因(HVG,highly variable genes)。HVG的鉴定可以大大加速下游分析速度以及都聚类结果产生重大影响,算法包括BASiCS, Brennecke, scLVM, scran, scVEGs和Seurat等。不同的算法运行时间及聚类结果大相径庭。有时候我们需要整合不同来源或者平台的单细胞数据,就会涉及批次效应的处理,目前常见的算法包括Harmony,Seurat,Scanorama,deepMNN和scGen等。
特征提取之后,降维算法的选择也多种多样,包括第一轮的PCA(principal component analysis)及第二轮的t-SNE (t-distributed stochastic neighbor embedding) 或者UMAP(Uniform Manifold Approximation and Projection)降维可视化。降维之后可以进行细胞注释,主要包括三个步骤:自动注释、人工校验、实验验证。自动注释是根据已知细胞类型的marker基因表达情况来注释细胞,常见的注释方法有Seurat, scmap, SingleR, CHETAH, SingleCellNet, scID, Garnett, SCINA, CP和RPC。但这种方法无法注释罕见或者新的细胞类型,以及容易混淆相近的细胞。所以第二步需要进行人工核验。通过挖掘现有文献的信息,结合细胞群体的功能等特征进行校验细胞类型注释是否正确。最后就是通过实验验证,一般包括免疫荧光及免疫组化来看细胞群体是否真实存在。
高级分析
细胞注释完成之后,就需要对感兴趣的细胞群体或者不同处理进行探索性分析,差异基因(DEG)及功能富集(GO/KEGG)是最常见的分析。GSVA也常用来评估造成表型差异的通路原因。SCENIC可以用来推测不同细胞群体中富集的转录因子(TF)。拟时间分析(Pseudo-time analysis)用来推断细胞发育轨迹及可能存在的隐藏状态。细胞互作(cell-cell communication)则可以分析不同群体之间的调节情况。此外,联合其他不同组学,则可以探索更多可能的分析。
参考文献
除非注明,文章均为原创,转载请以链接形式标注本文地址
本文地址:http://colorfulbiolife.com/omics/scrna/%e5%85%85%e5%88%86%e5%88%a9%e7%94%a8scrna-seq%e6%95%b0%e6%8d%ae/