





Seurat分析-基础篇
Seurat是一种用于单细胞RNA测序(scRNA-seq)数据分析的工具。它的主要目标是揭示单个细胞水平上基因表达的差异,帮助研究者理解细胞群体的异质性和特征。由美国布莱德学院(Brigham and Women’s Hospital)的生物信息学家Rahul Satija及其合作者开发的。Rahul Satija是生物信息学和计算生物学领域的专家,他的研究重点包括单细胞RNA测序数据分析以及细胞群体的生物学异质性研究。
Seurat最初于2015年首次发布,并且在之后的版本中经过持续的更新和改进。这个工具在单细胞生物学研究中得到了广泛的应用,为科学家们提供了强大的工具来深入了解单个细胞的基因表达和细胞群体的异质性。目前,Seurat已经发布到V5版本。
安装
# 通过CRAN安装
# Enter commands in R (or R studio, if installed)
install.packages('Seurat')
library(Seurat)
#安装以下R包会提高性能,非必须
setRepositories(ind = 1:3, addURLs = c('https://satijalab.r-universe.dev', 'https://bnprks.r-universe.dev/'))
install.packages(c("BPCells", "presto", "glmGamPoi"))
#安装以下R包,拓展用法
# Install the remotes package
if (!requireNamespace("remotes", quietly = TRUE)) {
install.packages("remotes")
}
install.packages('Signac')
remotes::install_github("satijalab/seurat-data", quiet = TRUE)
remotes::install_github("satijalab/azimuth", quiet = TRUE)
remotes::install_github("satijalab/seurat-wrappers", quiet = TRUE)
#此外建议大家安装ggplot2及ggsci
install.packages('ggplot2')
install.packages('ggsci')如果想安装更早版本的seurat,采用下面的方法:
# 安装Seurat4
remotes::install_version("SeuratObject", "4.1.4", repos = c("https://satijalab.r-universe.dev", getOption("repos")))
remotes::install_version("Seurat", "4.4.0", repos = c("https://satijalab.r-universe.dev", getOption("repos")))
#安装Seurat2-3
remotes::install_version(package = 'Seurat', version = package_version('X.X.X'))数据准备
前面cell ranger部分提到,10x genomics分析完成之后在filtered_feature_bc_matrix目录下包含了三个文件,依次表示鉴定到的barcodes,genes以及以三列形式存储的表达值(对于稀疏矩阵更友好)。我们从这里下载一套示例数据解压之后如下图所示。这是一套由10x genomics提供的外周血(PBMC)单细胞数据,共包含2700个细胞。

library(dplyr)
library(Seurat)
library(patchwork)
# Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "/path/to/filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
# pbmc.data为原始表达矩阵;所以如果是诸如Smart-seq2或者其他平台的单细胞数据,可以预先构建表达矩阵之后使用CreateSeuratObject构建Seurat对象
# min.cells = 3: 至少在3个细胞中表达的基因被保留
# min.features = 200: 至少表达200个基因以上的细胞被保留
pbmc
## An object of class Seurat
## 13714 features across 2700 samples within 1 assay
## Active assay: RNA (13714 features, 0 variable features)
## 1 layer present: counts
pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
## 3 x 30 sparse Matrix of class "dgCMatrix"
##
## CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5
## TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . .
## MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . .
# .表示表达值为0,通过这种方式降低存储使用数据预处理
Seurat提供了数据预处理的功能,包括基因表达的标准化、归一化和去噪。这有助于确保不同样本之间的数据可比性。
Seurat常用的QC标准有以下两方面:
1.细胞中检测到的基因数:
裂解的细胞或者死细胞仅有少量基因被捕获,鉴定出有表达
双细胞(Cell doublets or multiplets)则明显包含大量的基因。当然,有专门的双细胞检测工具。
2.细胞中检测到的UMI数,和基因数类似
3.低质量/垂死细胞通常表现出广泛的线粒体污染,对于特定类型的细胞,如心肌细胞,本身线粒体基因表达比例高的时候需要注意阈值设定。
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# 细胞所有的信息储存在meta.data中
head(pbmc@meta.data, 5)
## orig.ident nCount_RNA nFeature_RNA percent.mt
## AAACATACAACCAC-1 pbmc3k 2419 779 3.0177759
## AAACATTGAGCTAC-1 pbmc3k 4903 1352 3.7935958
## AAACATTGATCAGC-1 pbmc3k 3147 1129 0.8897363
## AAACCGTGCTTCCG-1 pbmc3k 2639 960 1.7430845
## AAACCGTGTATGCG-1 pbmc3k 980 521 1.2244898
# 可以通过pbmc$percent.mt 直接访问某列meta信息VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
#小提琴图可视化质控指标(也可以可视化基因表达等其他信息)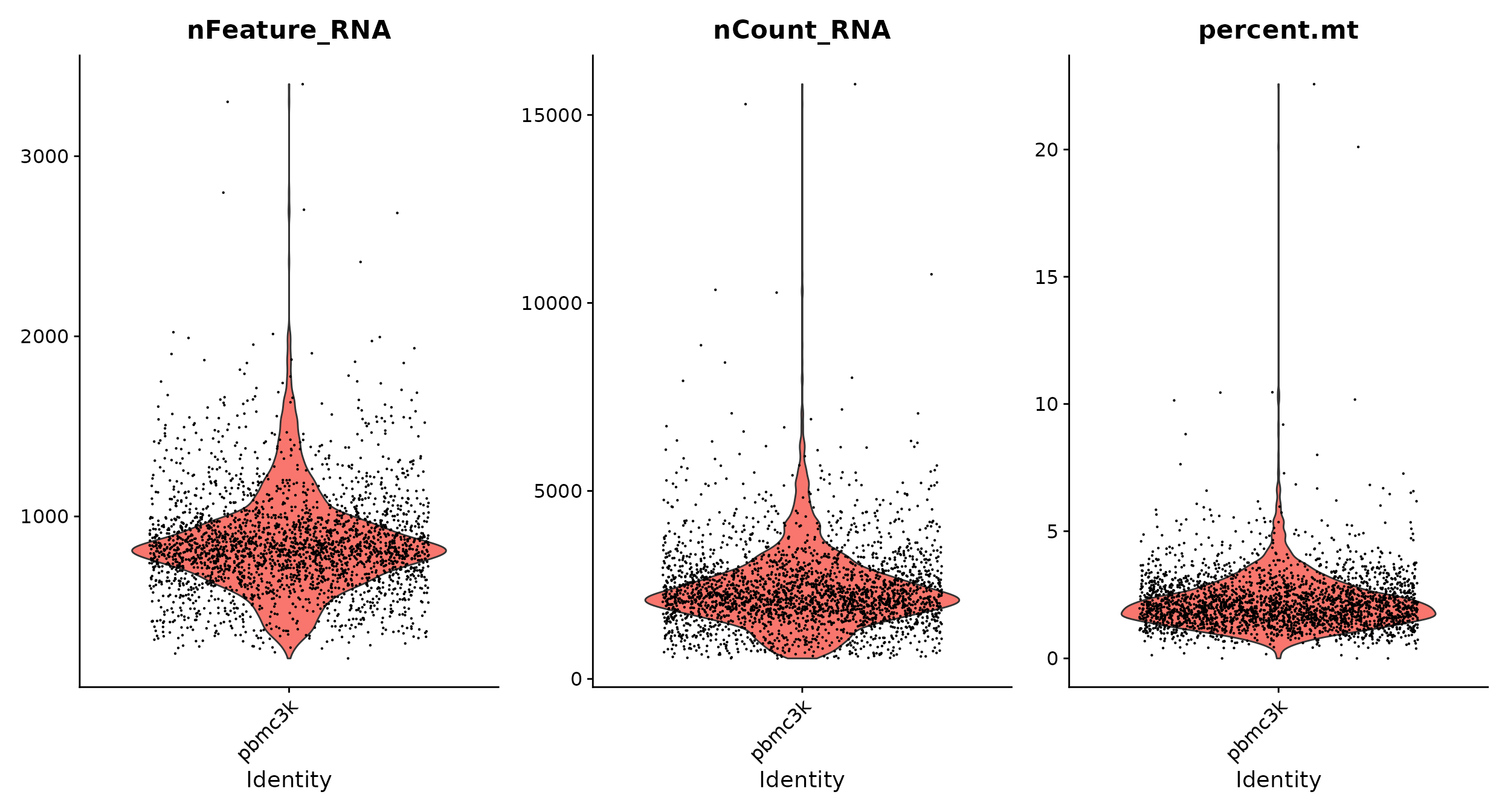
# FeatureScatter 用于展示两个特征(基因)的相对关系
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
# 使用subset可以过滤细胞,达到质控目的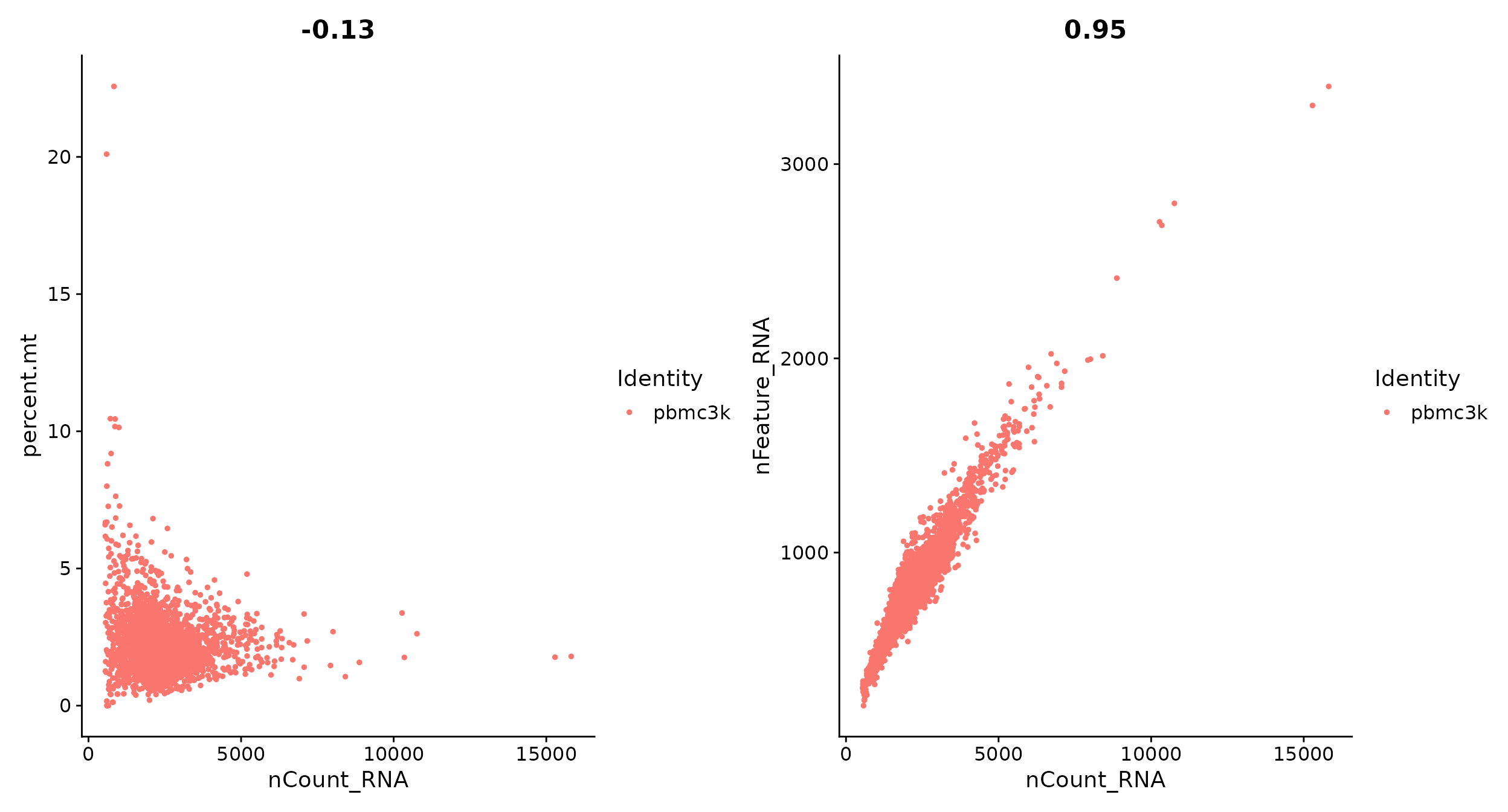
PCA降维
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
# 归一化表达值之后,乘以系数10000,然后进行对数变换,结果在pbmc[["RNA"]]$data
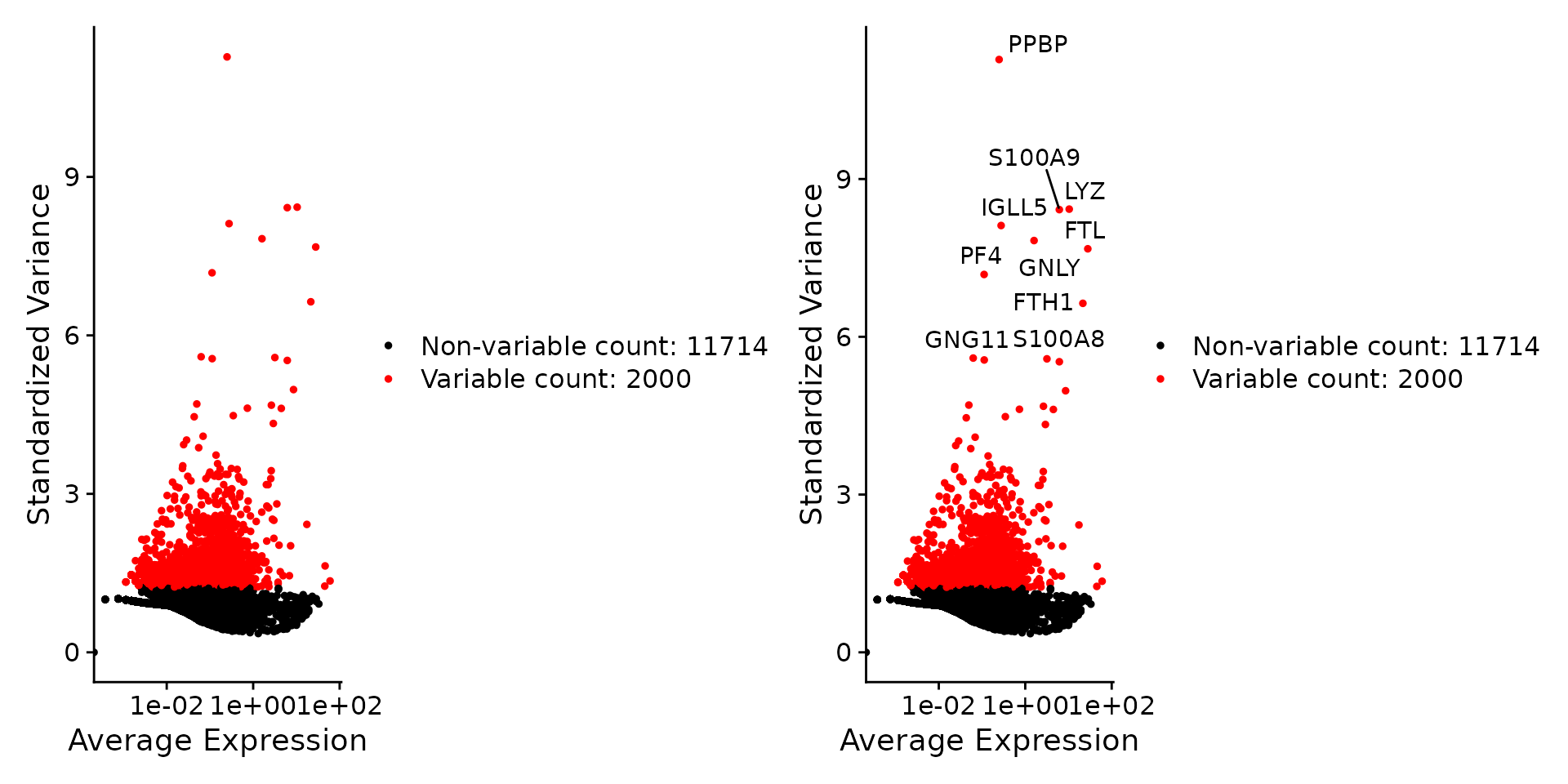
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# 高变基因鉴定;
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(pbmc), 10)
# plot variable features with and without labels
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2
计算Cell Cycle之前可以先下载用于预测细胞周期的基因,gene list来源于“Tirosh et al, 2015”
#计算Cell Cycle
s.genes <- cc.genes$s.genes
g2m.genes <- cc.genes$g2m.genes
# 小鼠
# s.genes = strsplit("Rrm2,Gmnn,Mcm4,Ccne2,Fen1,Cdc45,Casp8ap2,Dscc1,Pola1,Hells,Tyms,Slbp,Rpa2,Tipin,Msh2,Cdc6,Cdca7,Ung,Usp1,E2f8,Nasp,Blm,Exo1,Chaf1b,Brip1,Prim1,Rrm1,Rad51,Wdr76,Clspn,Gins2,Uhrf1,Mcm2,Ubr7,Rad51ap1,Mcm5,Mcm6,Rfc2,Dtl,Pcna",",")[[1]]
# g2m.genes = strsplit( "Cdc25c,Ckap2,G2e3,Cks2,Ect2,Kif20b,Hmmr,Dlgap5,Cdk1,Hmgb2,Smc4,Cenpa,Kif11,Anln,Rangap1,Tacc3,Tubb4b,Lbr,Hjurp,Kif2c,Tmpo,Cbx5,Cks1b,Cenpe,Ctcf,Ckap5,Ccnb2,Ndc80,Ncapd2,Kif23,Ttk,Anp32e,Gtse1,Aurkb,Cdca3,Cdc20,Cdca2,Psrc1,Gas2l3,Top2a,Nusap1,Cdca8,Nuf2,Nek2,Cenpf,Ckap2l,Birc5,Tpx2,Mki67,Ube2c,Aurka,Bub1" , ",")[[1]]
# 人
# s.genes = strsplit("MCM5,PCNA,TYMS,FEN1,MCM2,MCM4,RRM1,UNG,GINS2,MCM6,CDCA7,DTL,PRIM1,UHRF1,MLF1IP,HELLS,RFC2,RPA2,NASP,RAD51AP1,GMNN,WDR76,SLBP,CCNE2,UBR7,POLD3,MSH2,ATAD2,RAD51,RRM2,CDC45,CDC6,EXO1,TIPIN,DSCC1,BLM,CASP8AP2,USP1,CLSPN,POLA1,CHAF1B,BRIP1,E2F8",",")[[1]]
# g2m.genes = strsplit("HMGB2,CDK1,NUSAP1,UBE2C,BIRC5,TPX2,TOP2A,NDC80,CKS2,NUF2,CKS1B,MKI67,TMPO,CENPF,TACC3,FAM64A,SMC4,CCNB2,CKAP2L,CKAP2,AURKB,BUB1,KIF11,ANP32E,TUBB4B,GTSE1,KIF20B,HJURP,CDCA3,HN1,CDC20,TTK,CDC25C,KIF2C,RANGAP1,NCAPD2,DLGAP5,CDCA2,CDCA8,ECT2,KIF23,HMMR,AURKA,PSRC1,ANLN,LBR,CKAP5,CENPE,CTCF,NEK2,G2E3,GAS2L3,CBX5,CENPA",",")[[1]]
pbmc <- CellCycleScoring(pbmc, s.features = s.genes, g2m.features = g2m.genes, set.ident = TRUE)
# PCA
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, vars.to.regress = c("percent.mt","CC.Difference") )
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc), npcs = 30, verbose = TRUE)
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
DimPlot(pbmc, reduction = "pca") + NoLegend()
DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)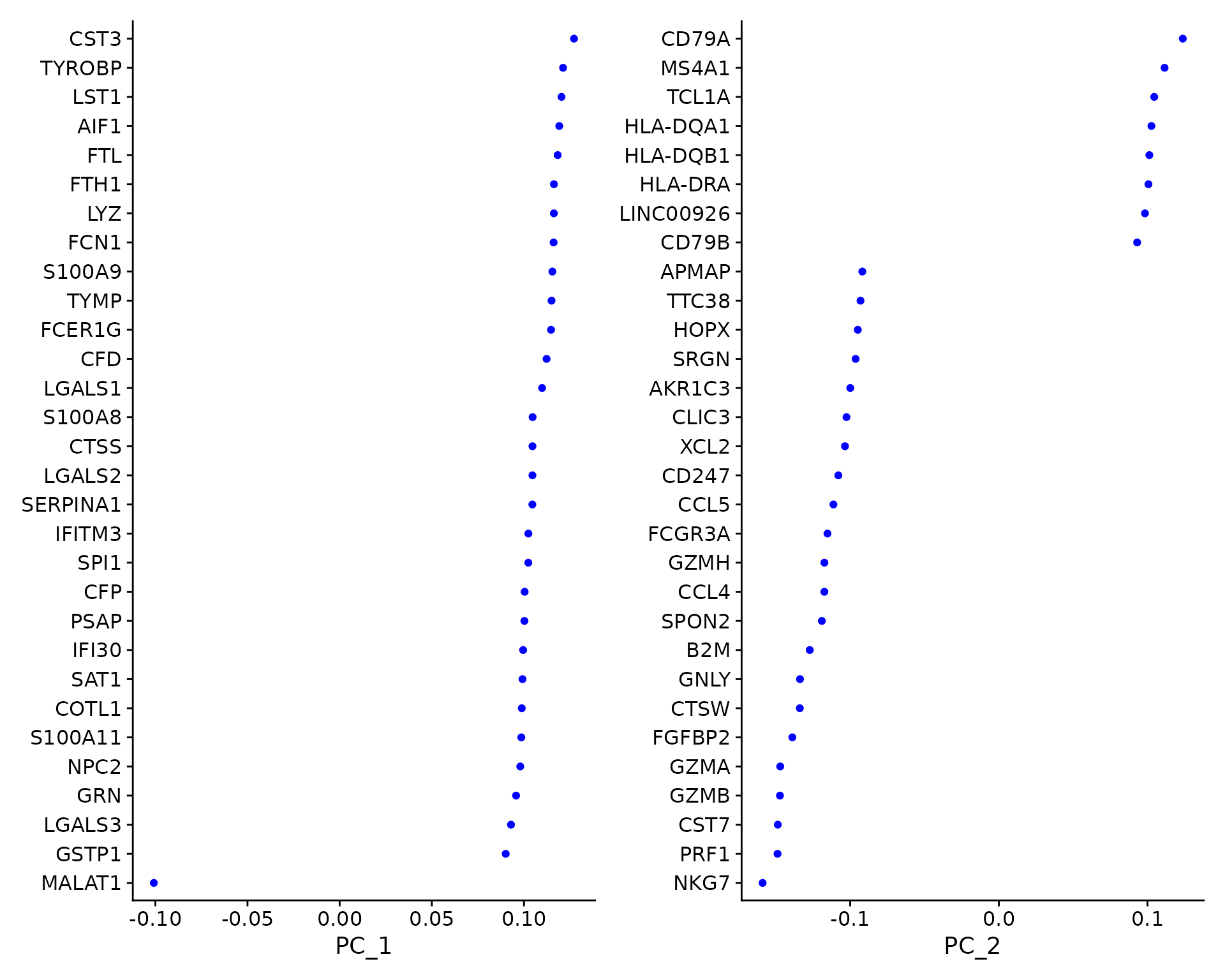
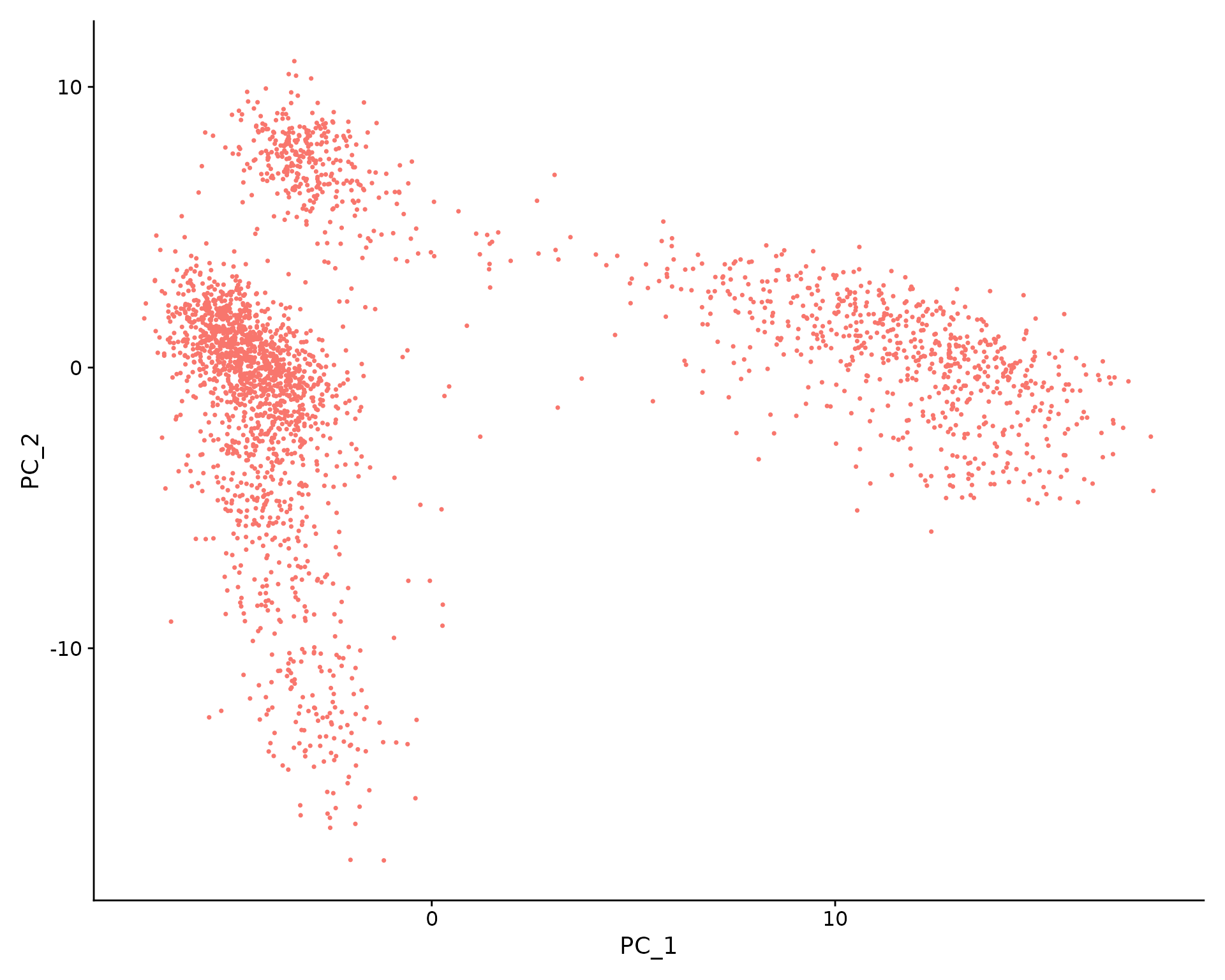

细胞聚类
Seurat基于PCA分数对细胞进行聚类,每个PC代表了一组“特征”,我们应该选择多少特征进行聚类呢?ElbowPlot根据每个主成分解释的方差百分比(ElbowPlot()函数)对主成分进行排序。“拐点”前的PC提供了大部分的真实信号。
ElbowPlot(pbmc)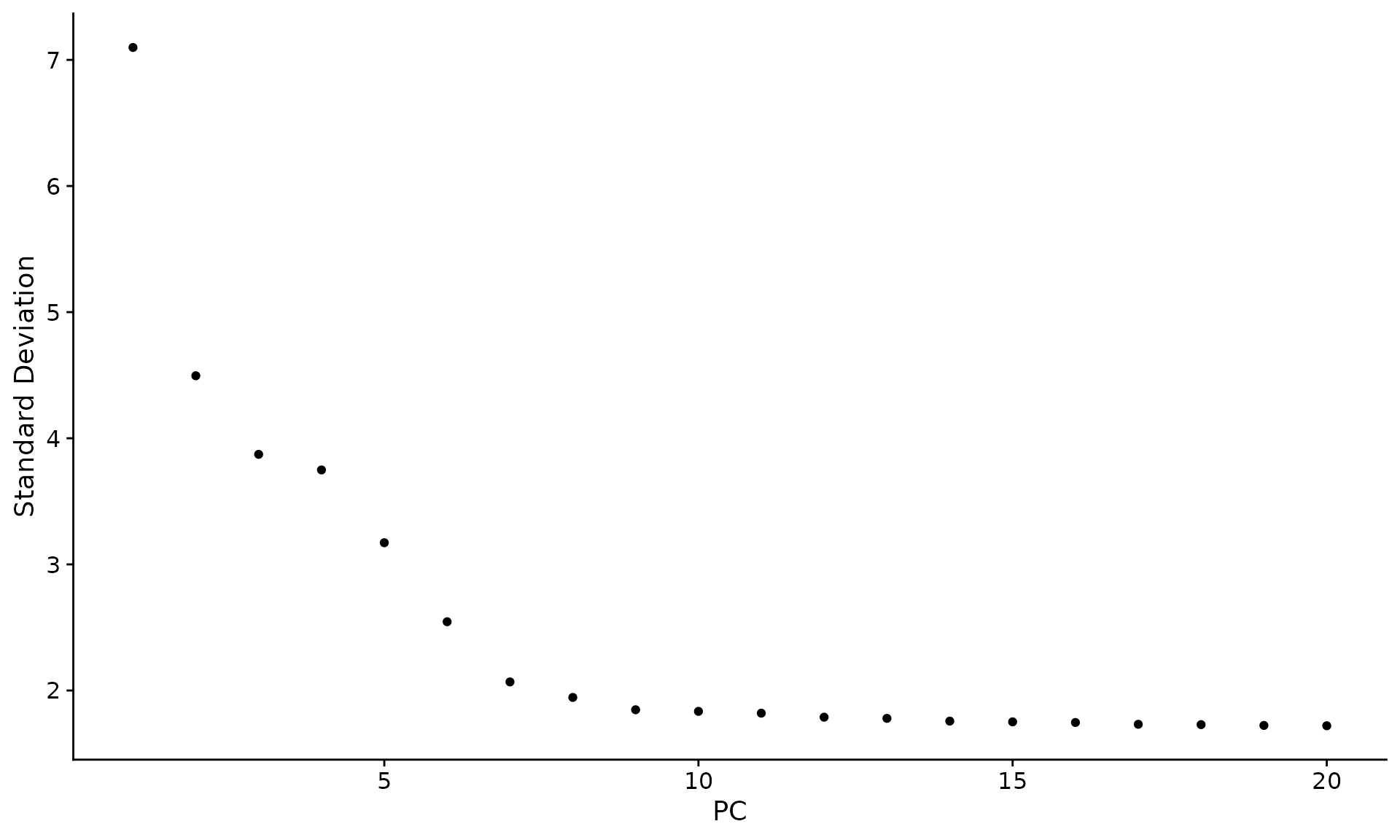
pbmc <- FindNeighbors(pbmc, dims = 1:10) # 计算细胞之间的两两距离及权重,构建KNN图
pbmc <- FindClusters(pbmc, resolution = 0.5) # 使用Louvain/SLM等算法对细胞进行迭代聚类,resolution表示聚类的精度,值越大,聚类数目越多
pbmc <- RunUMAP(pbmc, dims = 1:10) # 对数据进行可视化。图形分布不代表生物学状态。
saveRDS(pbmc, file = "pbmc_tutorial.rds") # 保存对象数据可视化
Seurat提供了丰富的数据可视化工具,包括降维可视化(如t-SNE和UMAP)和基因表达热图等,以帮助研究者更好地理解数据。
DimPlot(pbmc, reduction = "umap") # DimPlot展示特定分组信息,默认为聚类结果。可以通过DefaultIdents()修改默认类别
# split.by 按照值拆分UMAP图,可选meta.data中的值
# group.by 按照值展示不同的分组颜色,可选meta.data中的值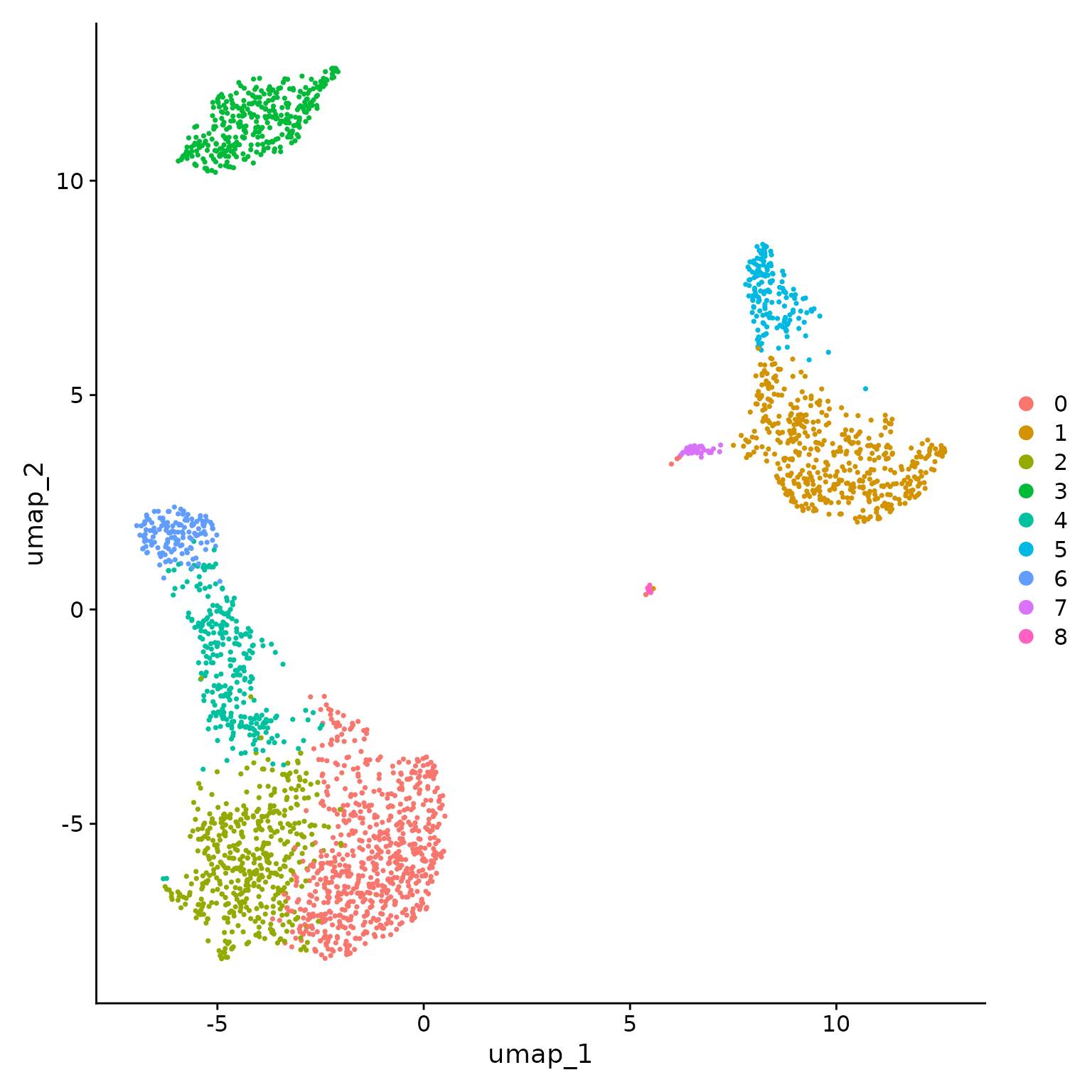
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE) #小提琴图可视化基因表达值,用counts值
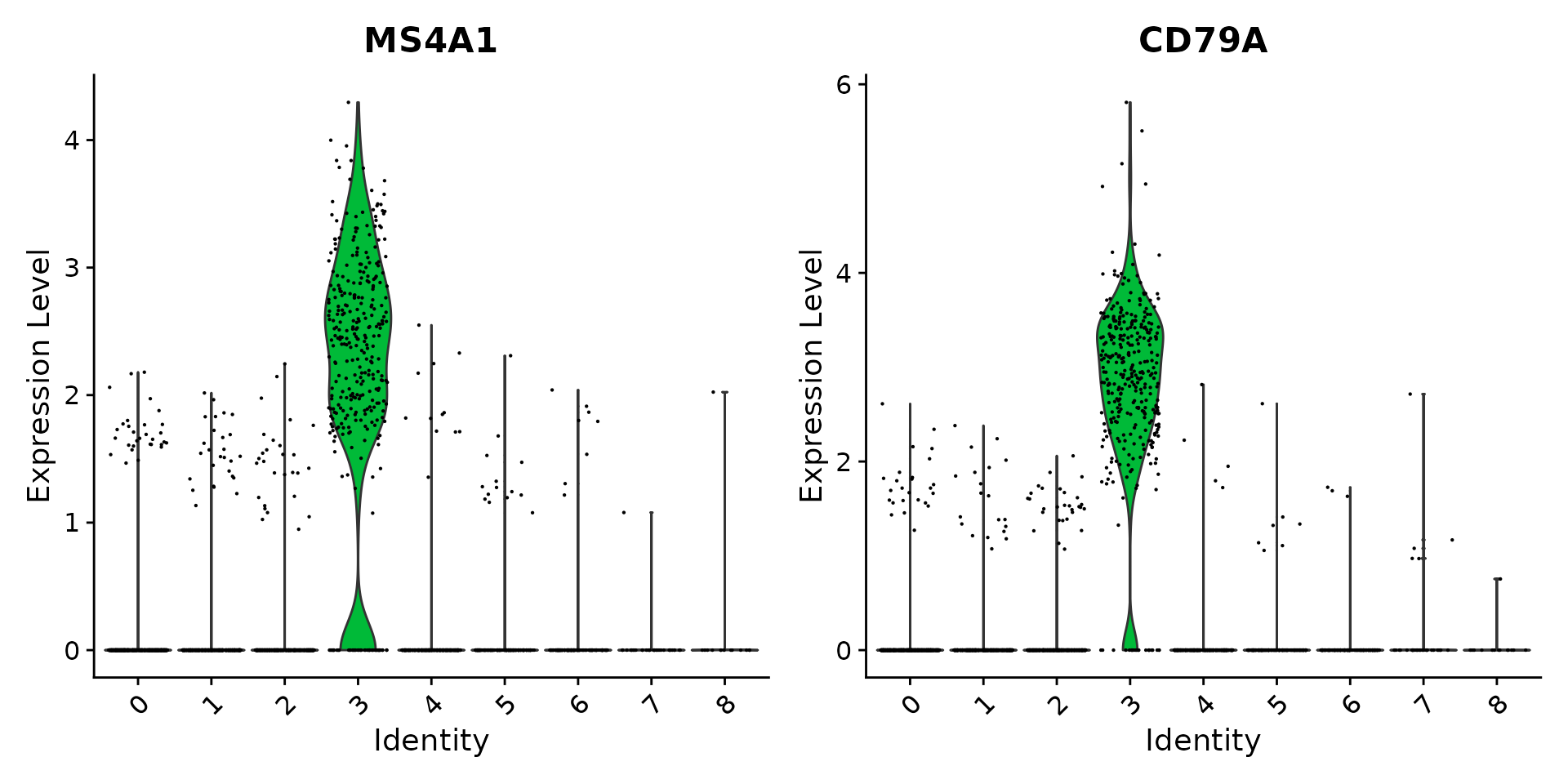
VlnPlot(pbmc, features = c("MS4A1", "CD79A")) #小提琴图可视化基因表达值,默认为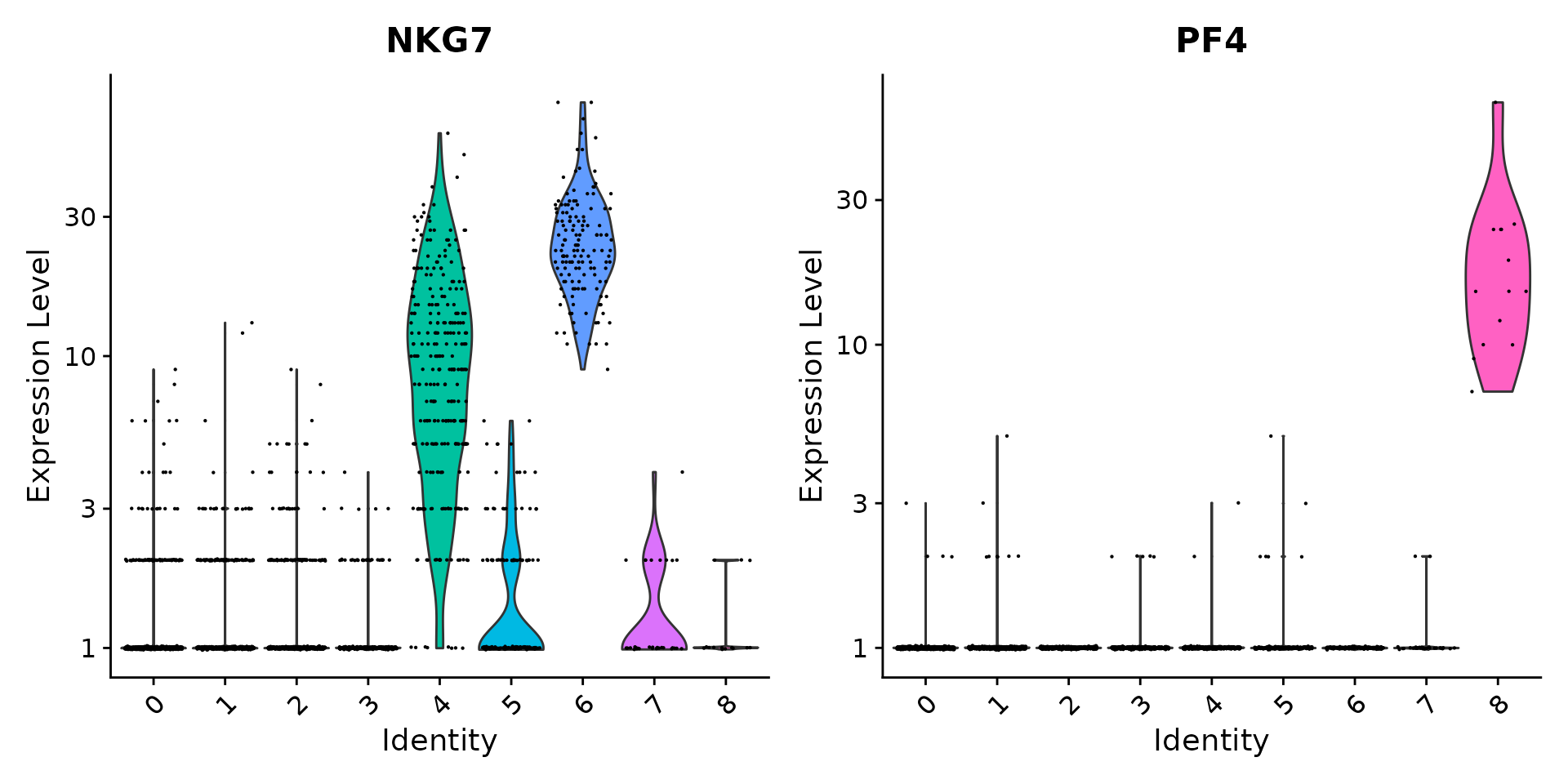
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A")) #FeaturePlot采用UMAP形式可视化基因表达情况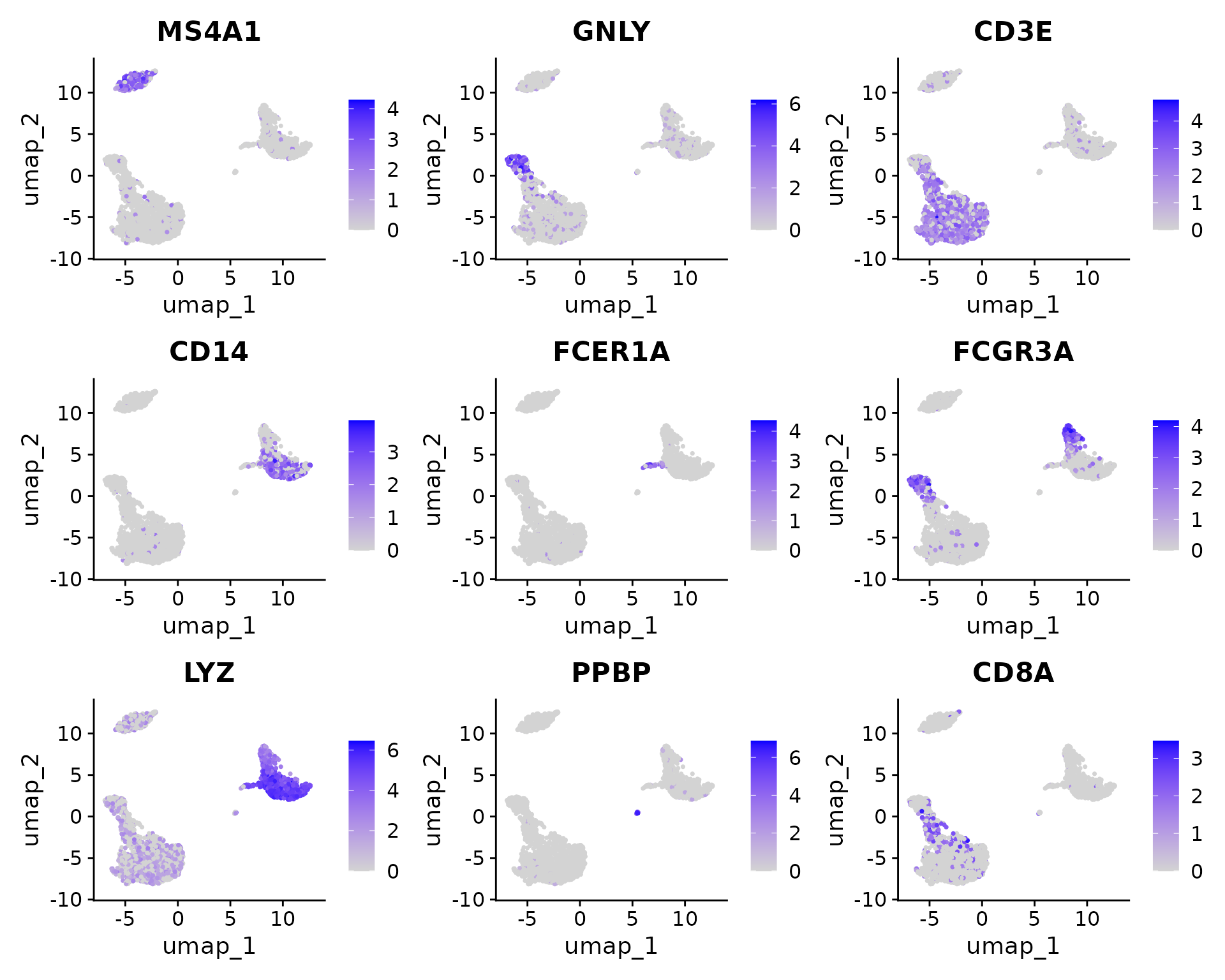
除非注明,文章均为原创,转载请以链接形式标注本文地址
本文地址:http://colorfulbiolife.com/omics/scrna/seurat%e5%88%86%e6%9e%90-%e5%9f%ba%e7%a1%80%e7%af%87/